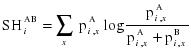
We present a web server implementation of the Sequence Harmony (SH) method previously introduced [Nucl Acids Res 34 6540]. The Sequence Harmony web server allows a quick selection of subtype-specific sites from a multiple alignment given a subfamily grouping, by scoring compositional differences, without imposing conservation. In addition, it maps the predicted sites directly onto a protein structure for display and manipulation. The Sequence Harmony Web Server can be accessed from: www.ibi.vu.nl/programs/SeqHarmWWW/ .
The Sequence Harmony method was previously developed and validated on a number of test sets. These mainly consisted of sets of site-specific mutations that were shown to induce a shift in function between the native and an alternative function. For pathway specificity for the SMAD proteins in the TGF-beta signaling pathway, SH selected 40 sites that included all 23 known specific sites in our newly assembled test set. For test sets assembled by others, SH performed equally well.
We demonstrate the use of the SH server using divergence between HIV populations in infected patients. The HIV sequence data was kindly provided by M. Navis & N. Kootstra, Clinical Viro- Immunology, Sanquin Research Amsterdam.
Table 1 shows the ranked list of selected sites, and Figure 2 shows selected sites in the crystal structure of the HIV capsid protein. Of the 26 sites selected by SH, 7 escape from B57 restricted cytotoxic lymphocites (CTL). Additional sites may relevant to the proliferation of HIV. Importantly, all known escape mutations for B57 in the part of the sequence analyzed, are selected. Other selected sites, therefore, are potentially interesting candidates for further investigation and (experimental) validation.
The main input is a multiple sequence alignment of the protein family and a subdivision into two groups, see Figure 1. Advanced features allow more control over the analysis and output, but the default settings usually suffice for a basic analysis. The SH cutoff can be adjusted between zero (allowing no compositional overlap) and one (allowing full overlap). A higher cutoff will lead to the selection of a larger number of sites. A reference sequence can be selected from the alignment to provide a reference numbering in the output tables. Additionally, a PDB identifier can be specified by its four-letter code, or uploaded, to visualise the selected sites in the protein structure. The Sequence Harmony between two groups (A and B) at position i is defined as follows:
Table 1 | (right) Output table of the Sequence Harmony analysis shows the 36 selected residues from the 422 residue alignment that are below the cutoff of 0.9. 'Rank' shows the length of sequentially consecutive ranges of selected sites.
Figure 1 | Input alignment (left) and web-form (right) of the Sequence Harmony Web Server, showing the input and settings for the analysis of the HIV alignment.
Figure 2 | (top) Low-harmony sites in the structure of the HIV capsid protein (1AFV and 2BU0), for the comparison of non-B57 vs. B57 patients.
|
This page was created using Emacs Last modified: Wed Nov 7 18:25:04 CET 2007 Back to Anton Feenstra Homepagefeenstra@chem.vu.nl |
 |